|
|
Az élőlények, bár tudományos alaposságú
meghatározásuk nem könnyű feladat, általában felismerhetőek néhány
alapvető jellegzetességük alapján. Az egyik legfontosabb
tulajdonságuk, hogy mozognak: másznak, futnak, repülnek, úsznak, vagy
éppen lassan növekednek formájuk megváltoztatásával egyidejűleg. A
mozgékonyság eredete után kutatva megfigyelhető, hogy az élő anyag
szerveződésének hierarchiájában egyre lejjebb és lejjebb hatolva
minden szinten megtaláljuk az arra jellemző mozgásformákat: az izmok
összehúzódnak, a testnedvek keringenek, a sejtek egymáshoz képest
eltolódhatnak vagy elvándorolhatnak, a sejtek belseje nem szűnő,
folyamatos áramlásban van. A sejten belül a sejtalkotók és a
makromolekulákból álló összetett struktúrák, komplexek világában is
találunk számos „mozgó alkatrészt”. Jelen írásomban az ezen képleteket
felépítő molekulák, melyek többségükben
fehérjék, belső mozgékonyságának jelentőségébe és vizsgálatába kívánok
rövid betekintést nyújtani.
A fehérjék húsz különböző fajta aminosav elvben
tetszőleges sorrendben és hosszúságig való összekapcsolódásával jönnek
létre. Az aminosavak sorrendje, szekvenciája jellemző az adott
fehérjemolekulára, és összetett módon meghatározza annak biológiai
feladatát. A klasszikus biokémiai nézet alapján a fehérje a szekvencia
által meghatározott, háromdimenziós térszerkezetet vesz fel, amely
célszerű alakjával és a meghatározott helyeken „felkínált” különböző
kémiai részletekkel, csoportokkal együtt határolja be és teszi
lehetővé az adott molekula biológiai szerepét.1
Ez a szerep lehet például (messze a teljesség igénye nélkül) más
molekulák átalakítása (enzimek), környezeti anyagok érzékelése
(receptorok), gének ki/be kapcsolása (transzkripciós faktorok), más
fehérjék aktivitásának szabályozása, a sejtek alakjának és
alakváltozásainak, mozgásainak meghatározása, illetve kivitelezése (a
sejtváz fehérjéi, illetve motorfehérjék). Minden fehérjére igaz
ugyanakkor, hogy „társas lény”, azaz mindig van (legalább) egy
partnermolekula (mely lehet fehérjetermészetű vagy egyéb), amelyen
keresztül a funkció megjelenik (ha más fehérje/nukleinsav/cukor vagy
egyéb molekula nem is, a minden fehérjét fiziológiás körülmények
között körülvevő víz szolgál partnerként, mint például a fagyásgátló
fehérjék esetében).
A háromdimenziós alak és a molekuláris funkció
kapcsolata az első fehérjeszerkezetek meghatározása, azaz a XX.
század 50-es évei óta jól ismert a kutatók számára, elviekben pedig
lényegesen régebbre (19. század vége) tekint vissza az Emil Fischer
által megfogalmazott kulcs–zár elmélet formájában. Eszerint a
partnermolekulák tökéletesen illeszkednek a fehérje által felkínált
bemélyedésbe, kötőzsebbe, és ez képezi a molekuláris felismerés és
funkció alapját. Ebbe a képbe merev, alakjukat egyáltalán nem
változtató fehérjemolekulák illenek bele.
A fehérjékről azonban az atomi és magasabb szintű
szerkezetük meghatározására kifejlesztett módszerekből tudjuk azt is,
hogy számos esetben többféle konformációval (egymásba könnyen
átalakuló térbeli szerkezettel) rendelkeznek, aminek sok biológiai
folyamatban nagy szerepe van. Jól ismert például, hogy a kalmodulin
nevű, általánosan előforduló Ca2+-szenzor fehérje kötőpartnerétől
függően többféle térszerkezetet vehet fel. A glikolízis egyik enzime,
a hexokináz ma már tankönyvi példája az átalakítandó molekula
(szubsztrát) kötése által indukált „bezáródásnak”. A sejtek felszínén
található, sejt–sejt és sejt–mátrix (sejtközötti állomány)
kapcsolatokért felelős, integrin nevű fehérjék a villamos
áramszedőjéhez hasonlóan rendelkeznek egy inaktív, behajlított és egy
aktív, nyújtott térszerkezettel, melyben a partnermolekula megkötésére
képesek. Ezen fehérjék esetében a különböző működési állapothoz
tartozó szerkezeteket külön kísérlet során, egymástól függetlenül
határozták meg, azaz statikus képeket vettek fel a molekula különböző
konformációiról. Természetesen a kutatókat az egyik szerkezetből a
másikba való átalakulási folyamat mechanizmusa és időskálája is
foglalkoztatja, mind elméleti, mind gyakorlati (például hatékonyabb
gyógyszerfejlesztés) szempontból, ez azonban
már jóval nehezebben vizsgálható kérdés.
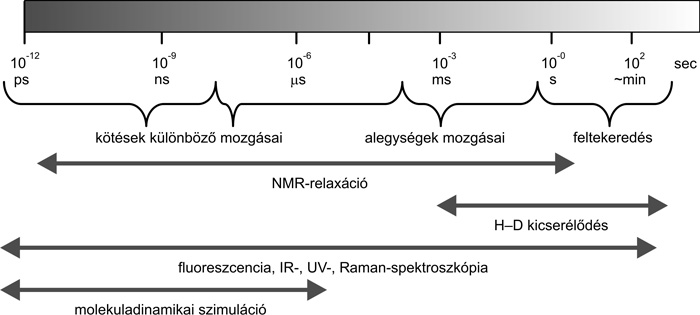
A ma elfogadott kép szerint a fehérjék igen széles
időskálán dinamikusak: mozgásaik a minden molekulára jellemző
kötésvibrációktól a feltekeredési folyamatokig (amikor a
„kitekeredett” fehérjelánc felveszi a biológiai funkcióhoz szükséges
háromdimenziós térszerkezetét) több mint tizennégy nagyságrendet
ölelnek fel, ami nagyobb különbség, mint ami egy szívdobbanás és egy
emberi élet hossza között van. Az eltérő időskálájú mozgásformák
különböző kísérleti (főleg spektroszkópiai) és elméleti módszerekkel
vizsgálhatóak (1. ábra).
A fehérjék térszerkezetének atomi szinten történő
vizsgálatára ma leginkább két módszert használnak a kutatók:
röntgenkrisztallográfiát és NMR-spektroszkópiát (NMR – nuclear
magnetic resonance, azaz mágneses magrezonancia) Előbbi kristályos
fázisban képes a molekula adott konformációjáról/konformációiról igen
pontos és részletgazdag, ám statikus képet adni. Az NMR-spektroszkópia
ugyanakkor oldatfázisban vizsgálja a molekulákat: itt a fehérjék a
kristállyal ellentétben nagy konformációs szabadsággal rendelkeznek,
következésképpen a mért spektroszkópiai paraméterek – legalábbis a
gyors mozgásokra vonatkozóan – a mintacsőben található konformációs
sokaság átlagát tükrözik. A biomolekulák térszerkezet-meghatározása
során rutinszerűen a hidrogénmagok távolságáról információt adó
nukleáris Overhauser-effektust (NOE) használjuk fel, amely a fentiek
értelmében, flexibilis molekulákról lévén szó, egy-egy adott
atomtávolság esetében igen bizonytalan (átlag)érték, azaz nem
feltétlenül igaz az, hogy az összes mért távolság egyszerre teljesül
egy adott molekula esetében. Elegendően sok ilyen atomi távolság
felhasználásával azonban kaphatunk egy jól jellemezhető konformert. A
ma rutinszerűen alkalmazott térszerkezet-meghatározó eljárások során
számos térszerkezetet generálunk, majd ezek közül kiválasztjuk azokat,
amelyek egymáshoz hasonlóak, és lehetőleg minden NOE-alapú távolság
jellegű kényszerfeltételt kielégítenek. Ezek a szerkezetcsaládok, bár
természetesen a szerkezetre/működésre vonatkozóan értékes biokémiai
adatokat szolgáltatnak, a röntgendiffrakció precizitását a NOE-adatok
pontatlansága miatt nem képesek elérni, ugyanakkor a számítás módja
miatt nem tükrözi a molekula valós dinamikáját sem.
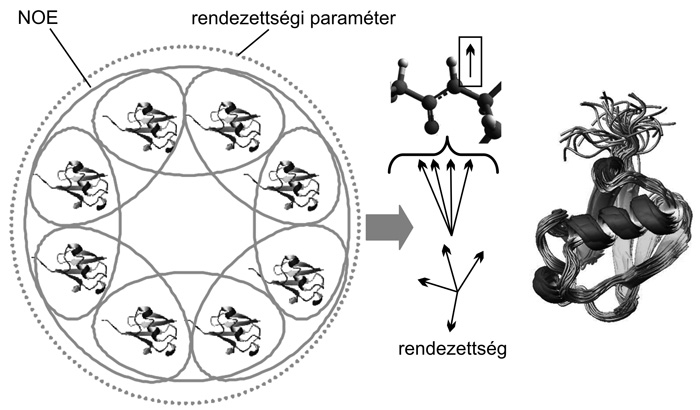
A modern NMR-spektroszkópia a NOE-adatok mellett
számos egyéb paraméter mérésére,
meghatározására alkalmas, és ezen paraméterek némelyike közvetlen
kapcsolatban áll a molekulák belső dinamikájával. A tipikusan
heteroatomok (15N, 13C izotópok) relaxációs tulajdonságai (gerjesztés
után az alapállapotba való visszatérés mikéntje) alapján számolt
rendezettségi paraméterek (a szakirodalomban elterjedt nevükön S2
értékek) az adott atom és a kapcsolódó hidrogén által meghatározott
kötésvektor piko- és nanoszekundum időskálájú mozgásának
kiterjedtségéről adnak képet (a
2. ábra jobb oldalán). Mivel ez az
információ viszonylag könnyen lefordítható térbeli heterogenitásra,
néhány éve megjelentek olyan szerkezetfinomító számítási eljárások,
amelyek explicit módon figyelembe tudják venni az S2 értékeket. Ezek
közül az egyik legújabb módszer a MUMO (Minimal Underrestraining
Minimal Overrestraining, Richter et al, 2007), amely esetében
lehetőség van arra, hogy a különböző típusú paramétereket más-más
méretű sokaságon vegyük figyelembe a szerkezetfinomítás során. A
|
|
|
bemutatott példán (2. ábra) nyolc molekula
párhuzamos molekuladinamikai szimulációja során az NOE-alapú
kényszerfeltételeket páronként, míg a rendezettségi paramétereket mind
a nyolc példányra egyidejűleg próbáljuk meg teljesíteni. Ezáltal
megfelelünk annak a kívánalomnak, hogy a NOE-adatokat kettőnél nagyobb
sokaságra nem átlagoljuk, tehát nem engedjük meg a konformerek túlzott
eltávolodását egymástól (elkerülve a túlillesztést vagy túlságosan
laza megkötést), egyúttal a nyolc példány elegendő teret biztosít a
rendezettségi paraméterek által jellemzett heterogenitás
megjelenésének (minimális alulillesztés, illetve nem túlságosan szoros
megkötés). Egy így előállított konformációs sokaságtól azt várjuk,
hogy az egyes konformerek közötti különbségek a molekula valós
dinamikájából adódnak, tehát egy időbeli jellemző biológiailag
releváns térbeli leírását hoztuk létre.
A kapott, úgynevezett dinamikus konformációs
sokaságok alkalmasak tehát arra, hogy az elvont, számszerűen megkapott
rendezettségi paramétereket „lefordítsuk” a teljes molekulában
megvalósuló mozgások „pillanatképekkel” jól jellemezhető szintjére,
némileg hasonlóan ahhoz, amikor például a ló vágtázó mozgását gyors
egymásutánban felvett állóképek segítségével jellemezzük. Az
alábbiakban két, kutatócsoportunkban vizsgált fehérjét mutatok be
példaként.
A leírói által Tc5b-nek keresztelt molekula az
egyik legkisebb olyan fehérje, amely vizes oldatban jól meghatározott
térszerkezetet vesz fel (Neidigh et al, 2002). (A hozzá hasonló méretű
fehérjék általában nagyszámú, egymástól jelentősen eltérő
konformációval jellemezhetőek.) Éppen ezért jól használható modell a
nagyobb fehérjék stabilitásának és dinamikájának megértéséhez
(gyakorlati jelentőségét pedig az mutatja, hogy egy származéka 2008
óta cukorbetegség gyógyítására forgalomba hozott gyógyszer).
Csoportunkban sikerült egyetlen apró kémiai módosítással az eredeti
Tc5b egy továbbstabilizált változatát létrehozni (Hudáky et al, 2008).
A Tc5b és az előállított Tc6b térszerkezete kismértékben különbözik,
ami együtt jár a belső dinamika megváltozásával is. A Tc6b esetében
megfigyelhető, hogy a belső mozgások valamivel egyenletesebbek és a
molekula végei kisebb mértékű heterogenitást mutatnak, mint az eredeti
molekula esetében (3. ábra). A stabilitás, a térszerkezet és a
dinamika megváltozása tehát elválaszthatatlan egymástól.
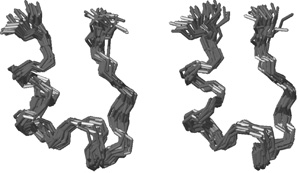
3. ábra • Az eredeti Tc5b (jobbra) és
stabilizált változatának (balra) dinamikus sokaságai a molekulagerinc
sematikus ábrázolásával
Mint arról korábban már szó volt, a fehérjék mindig
más molekulákkal kölcsönhatásba lépve fejtik ki biológiai szerepüket.
Az általunk vizsgált kisméretű proteázinhibitorok nevüknek megfelelően
fehérjéket lebontó enzimek gátlása révén hatnak. Az SGCI rövidítéssel
jelölt fehérje kísérleteink alapján igen dinamikusnak mutatkozott: a
benne stabilizáló szerepet betöltő három diszulfidhíd (a molekulalánc
mentén távoli kénatomjai között létrejövő kovalens kapcsolat)
jelenléte ellenére a kapott rendezettségi paraméterek jelentősen
elmaradnak a nagyobb fehérjékre általában jellemző értékektől (Szenthe
et al, 2004). Ezzel első ránézésre nehezen hozható összhangba, hogy az
SGCI kiváló enzimgátló, hiszen ezt a funkciót tipikusan merevebb
fehérjéknek és egyéb (például jóval kisebb gyógyszer-) molekuláknak
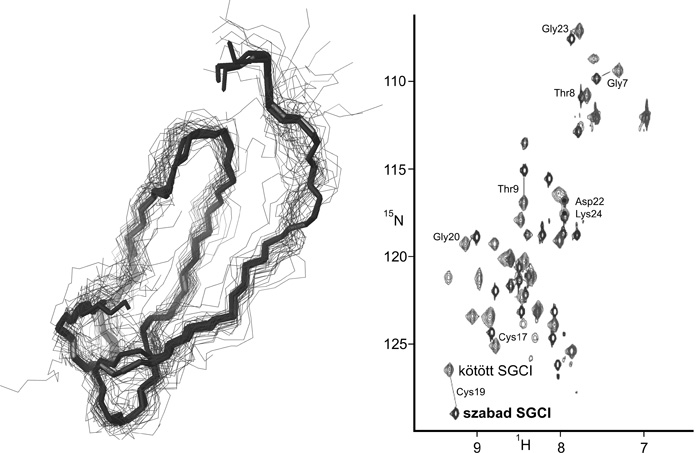
tulajdonítják. Az SGCI enzimmel való kölcsönhatását
NMR-spektroszkópiával vizsgálva az inhibitor egészében látunk
változásokat, nem csupán a partnermolekulával közvetlenül
kölcsönhatásba lépő, viszonylag kis kiterjedésű részletében (Gáspári
et al, 2006). Az SGCI dinamikus sokaságát előállítva fény derült arra,
hogy az igen dinamikus molekulának szabad
formában felvett konformációi között olyanok is vannak, amelyek igen
közel esnek a kötött állapotban megvalósuló szerkezethez. Bár az
észlelt szerkezeti változások nem látványosan nagyok, ez a
megfigyelés, összevetve az NMR-spektroszkópiával észlelt nagyobb
léptékű változásokkal azt sugallja, hogy az enzim nem „a maga képére
formálja”, hanem mintegy csupán kiválasztja a megvalósuló sokaságból a
kötött állapotnak megfelelő formát, egyúttal megváltoztatva az egyes
konformációk egymásba alakulásának egyensúlyát. A megfigyelt, teljes
molekulára kiterjedő változások eszerint tehát nem térbeli, hanem
elsősorban dinamikai jellegűek (4.
ábra). Egyéb adatokkal együtt az a kép bontakozik ki,
hogy a molekuláris partnerek között nem csupán geometriai, hanem
mozgékonysági megfelelés, illeszkedés is megvalósul.
A 19. század elején a szerkezeti biológia egyik
nagy kihívása a biomolekulák belső dinamikájának feltárása és a
biológiai folyamatokban való szerepének tisztázása. Jelenleg képesek
vagyunk ezt a dinamikát elsősorban NMR-spektroszkópiai módszerekkel
atomi szinten feltárni, és ma már vannak látványos példák arra, hogy
ez a dinamika hogyan játszik szerepet például enzimek működésében
(Eisenmesser et al., 2005). A dinamika kézzelfogható megjelenítésével,
dinamikus sokaságok előállításával pedig közelebb kerülünk a fehérjék
valódi természetének leírásához és megértéséhez. Ha megismerjük a
fehérjék szóló- és társastáncainak koreográfiáját, valóra válhat az
élőlényekben sok tér- és időbeli tartományban jelenlévő mozgás
gyökereinek feltárása.
Kulcsszavak: fehérje, belső dinamika, NMR-spektroszkópia,
rendezettségi paraméter, dinamikus sokaság, enzimműködés
IRODALOM
Eisenmesser, Elan – Millet, O. –
Labeikovsky, W. et al. (2005): Intrinsic Dynamics of an Enzyme
Underlies Catalysis. Nature. 438, 117–121.
Gáspári Zoltán – Szenthe B. – Patthy A. et
al. (2006): Local Inding with Globally Distributed Changes in a Small
Protease Inhibitor upon Enzyme Binding. The FEBS Journal. 273,
1831–1842.
Hudáky Péter – Stráner P. – Farkas V. et
al. (2008): Cooperation between a Salt Bridge and the Hydrophobic Core
Triggers Fold Stabilization in a Trp-cage Miniprotein. Biochemistry.
47, 1007–1016.
Neidigh, Jonathan W. – Fesinmeyer, R. M. –
Andersen, N. H. (2002): Designing a 20-Residue Protein. Nature
Structural Biology. 9, 425–430.
Richter, Barbara – Gsponer, J. – Várnai P.
et al. (2007): The MUMO (Minimal Under-restraing Minimal
Over-restraining) Method for the Determination of Native State
Ensembles Of Proteins. Journal of biomolecular NMR. 37, 117–135.
Szenthe Borbála – Gáspári Z. – Nagy A. et
al. (2004): Same Fold with Different Mobility: Backbone Dynamics of
Small Protease Inhibitors from the Desert Locust, Schistocerca
gregaria. Biochemistry. 43, 3376–3384.
1 Az utóbbi években
felfedezett és egyre jelentősebbnek tartott, önmagukban határozott
térszerkezettel nem rendelkező, ún. funkcionálisan rendezetlen
fehérjékről Tompa Péter írása szól.
<
|
|