|
|
A mágneses magrezonancia (nuclear magnetic
resonance – NMR) spektroszkópia napjainkra a kémiai szerkezetkutatás
valószínűleg leghatékonyabb és ennek megfelelően legelterjedtebb
eszköze (Sohár, 1976, 1983a; Hore 1995).
Még száz éve sincs, hogy Wolfgang Pauli azon
feltételezése (1925), amely szerint az atommagokban mágneses tér
hatására paramágneses momentum indukálódik, megalapozta e fiatal, ám
roppant jelentőségűvé fejlődött új tudományágat. A paramágneses
momentum vektora – a spin – úgy viselkedik, mint egy parányi
iránytű, amelyet a mágneses tér a maga irányába kényszerít. A spin
kvantált természetű, azaz csak meghatározott értékei, s ennek
megfelelően az atommagoknak diszkrét mágneses energiaállapotai
lehetségesek. A valamely térirányban mérhető m mágneses momentum
gh/(2π)-nek csak fél-egész vagy egész számú többszöröse, mgh/(2π)
lehet. Itt a g konstans, az ún. giromágneses tényező, anyagi
jellemző, h pedig a Planck-állandó. Az m futószám az I, I-1,
I-2,…-I, összesen 2I+1 értéket veheti fel, ahol I a spinkvantumszám.
Minden atommagfajta – izotóp –, amelyre I nem zérus, „mágneses mag”:
mágneses térben képes a rezonanciaabszorpcióra. (I = 0 csak azokra
az izotópokra áll fenn, amelyekben a protonok és neutronok száma
egyaránt páros.) Ha I = ½, akkor két állapot van: m = ½ és m = −½,
ha I = 1, akkor a kvantumállapotok száma 3 (m = 1, 0 vagy −1), és
így tovább. A legegyszerűbb esetben tehát két állapot lehetséges: a
kedvezőbb, amikor a spinvektor térirányú, de ezzel ellentétes,
nagyobb energiájú beállásra is gerjeszthetjük erre alkalmas
energiájú, rádiófrekvenciás (RF-) sugárzás elnyeletésével. Az alap-
és gerjesztett állapotok energiakülönbsége adott: a folytonos
RF-sugárzásnak csak az ennek megfelelő frekvenciájú komponensét
képesek – tehát rezonanciaszerűen és csak mágneses térben –
abszorbeálni az atommagok: innen a jelenség elnevezése. A frekvencia
függvényében ábrázolt energiaabszorpciós jelek képezik az
NMR-spektrumot. A rezonanciafrekvencia anyagi jellemző, minden
izotópra más, de arányos a mágneses tér B0 erősségével. Az eddigiek
értelmében minden mag, minden izotóp NMR-spektruma egyetlen
elnyelési maximumot tartalmaz.
A múlt század húszas éveiben feltételezett rezonanciajelenség
kísérleti bizonyítására két évtizedet kellett várni. A mágneses
kvantumállapotok között csak addig lehet átmeneteket létrehozni
(energiaelnyeléssel a kisebb energiájú állapotból a nagyobb
energiájúba gerjeszteni), amíg a magok száma a két állapotban
azonossá válik (a magrendszer telítődik), a betöltöttségkülönbség
kiegyenlítődik (a termodinamikai törvények értelmében a kisebb
energiájú állapotban több mag tartózkodik, de csak a többletet lehet
gerjeszteni). Mivel a betöltöttség- és az energiakülönbség az
állapotok között roppant kicsiny, a kevés energia rövid ideig tartó
elnyelését nagyon nehéz kimutatni. Például, ha szobahőmérsékleten,
egyensúlyban a gerjesztett állapotú 1H-magok száma 106, akkor
alapállapotban mindössze 66 maggal van több. Csak 1945-ben sikerült
két amerikai, Felix Bloch és Edward Mills Purcell vezette
kutatócsoportoknak a rezonanciát kísérletileg igazolniuk. Mindkét
csoport a hidrogénmag RF-elnyelését detektálta, amelynek
„NMR-érzékenysége” az összes mágneses magok közül a legnagyobb. Az
NMR-érzékenység nagyságrendekkel tér el a különböző magfajtákra, és
elsősorban a természetes izotópgyakoriságtól, továbbá a mágneses
momentum és I nagyságától függ. Az 1H izotóp például közel 100%-a a
természetes izotópkeveréknek, míg például a 13C mag csak kb. 1%-ban
fordul elő a mágnesesen inaktív 12C (I = 0) izotópja mellett. Ezért,
és a kisebb mágneses momentum miatt a 13C izotóp rezonanciájának
kimutatása (az általa elnyelt energia nagysága miatt) mintegy három
nagyságrenddel nagyobb érzékenységet követel. Azon atommagoknak,
amelyek I spinkvantumszáma nem ½, elektromos kvadrupólmomentumuk
van, amely a rezonanciajelek nagymértékű kiszélesedését okozza, s
ezzel igen nehézzé teszi detektálásukat. Ezért az ½ spinű izotópok,
közöttük az 1H jelének detektálása a legkönnyebb.
Mindaz, amiről eddig szó volt, a kvantumelmélet és
a kísérleti fizika tárgykörébe tartozik, s az említett
méréstechnikai problémák miatt az 1930–1940-es években sokan már
„temették” az NMR-módszert, mondván, hogy csak elméleti jelentősége
van, gyakorlati haszna alig, amivel nincsenek arányban a kísérleti
nehézségek és költségek. A múlt század második felének kezdete táján
azonban a kémia is felfedezte a maga számára az NMR-t, és hamarosan
a kémiai szerkezetkutatás számára ma már nélkülözhetetlen módszerré
fejlődött, szinte határtalan, napjainkig is hihetetlen tempóban
bővülő alkalmazási lehetőségek birtokába juttatva a vegyész
kutatókat és gyakorlati szakembereket.
A fordulat kulcsmomentuma a kémiai eltolódás
jelenségének felismerése volt. A fentiek szerint adott „külső”, B0
nagyságú mágneses térben minden atommagra egyetlen
rezonanciafrekvencia jellemző, amely csak a térerősségtől függ, és
azzal minden izotópra azonos arányban változik. Kitűnt azonban, hogy
a rezonanciafrekvencia, bár csak igen csekély mértékben, de a kémiai
környezettől is függ, vagyis az adott izotópra jellemző értéke
molekuláris kötelékben kissé megváltozik, s e változás mértéke a
kémiai eltolódás (d), amely tehát informál az illető mag molekuláris
környezetéről, azaz a kémiai szerkezetről. A változások igen
kicsinyek ugyan – a B0 értékéhez képest négy–hat nagyságrenddel
kisebbek (ezért praktikus okokból milliomodrészekben,
ppm-egységekben mérjük) –, de pontos mérésük esetén szinte
korlátlanul gazdag információforrást képviselnek a kémiai
szerkezetre vonatkozóan. A fentiek azt jelentik, hogy az
NMR-spektrum első közelítésben annyi jelből áll, ahány különböző
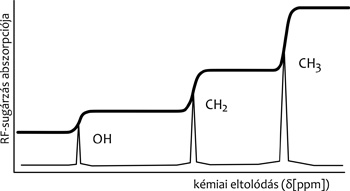
környezetben fordul elő a mért mag a vizsgált molekulában. Az 1.
ábrán a vizsgált vegyület 1H NMR spektruma például három jelből
áll a CH3, CH2 és gyűrűs CH csoportokbeli
hidrogéneknek megfelelően.
A kémiai eltolódás oka a molekulák elektronjainak a
mágneses térerőre gyakorolt hatása, ezért az elektronsűrűséget vagy
-eloszlást befolyásoló minden tényező a kémiai eltolódás jellegzetes
értékéhez, változásához vezet. Adott környezetben, funkciós
csoportban vagy kémiai kötésben előforduló magokat karakterisztikus
d-értéktartományok jellemzik, amelyek ún. korrelációs táblázatokba
foglalhatók, vagy empirikus egyenletekkel írhatók le. Ezzel lehetővé
válik egy adott mag, például hidrogén adott molekulabeli, kémiai
szerkezetbeli vagy szerkezeti elembeni kémiai eltolódásának jóslása,
s a mért d-értékekből ezek jelenlétének igazolása. A multinukleáris
NMR (ugyanazon mintában többféle, például 1H, 13C, 15N stb. mag
spektrumának mérése) megsokszorozza a szerkezetről árulkodó
információk számát.
1. ábra
Például az 1H NMR-spektrum lehetővé teszi a
telített, telítetlen és aromás csoportokbeli hidrogének
megkülönböztetését, sőt ezek számarányának meghatározását is egy
adott mintában. A rezonanciajelek intenzitása ugyanis az 1H
NMR-spektrumaiban igen jó közelítésben megadja a hozzájuk rendelhető
hidrogénatomok relatív számát (1. ábra). Ugyanezért
többkomponensű keverékek összetételét meg lehet határozni a
komponensektől származó jelek relatív intenzitása alapján. Kinetikai
mérések (valamely kémiai reakció időbeli lezajlásának követése)
lehetségesek a reagens vagy a termék jelintenzitás-változásának
(gyengülésének vagy erősödésének) időfüggő mérésével.
A 13C NMR-spektrum egyebek mellett a hidrogéneket
nem tartalmazó szerkezeti elemek (például karbonil- vagy
nitrilcsoport) az ún. téreffektus révén a térben egymás mozgását
gátló, akadályozó („zsúfolt”) csoportok kimutatását, a rendűségükben
eltérő (CH3, CH2, CH és kvaterner C), vagy hibridállapotukat
illetően különböző (telített, sp3, olefin, sp2 és acetilén típusú
sp) szénatomok jelenlétének igazolását (Sohár, 1976, 1983b; Wehrli –
Wirthlin, 1976; Breitmaier – Voelter, 1974). A háromtagú gyűrűs
vegyületek (ciklopropánok, oxiránok, tiránok és aziridinek) a
különleges, „hajlott” kötéseknek köszönhetően mind az 1H NMR, mind
pedig a 13C NMR segítségével biztonsággal felismerhetők. A 15N NMR
kémiai eltolódások például a nitro-, az amin- és amid NH-csoportok
jelenlétének bizonyítására adnak lehetőséget.
A fentiekben tárgyalt fő információfajta, a kémiai
eltolódás és a járulékos, jelintenzitásokból származó másik
információforrás mellett továbbiakat is nyerhetünk az
NMR-mérésekből. A kémiai eltolódással összemérhető fontosságú
információtípus a J csatolási állandó. A molekulákban egymáshoz
közeli atommagok spinjei – mágneses momentumai – megváltoztatják az
egymás körüli lokális mágneses teret, s vele az illető mag kémiai
eltolódását, mégpedig kvantumállapotuktól függően eltérő mértékben.
Egy ½ spinű „A” mag ezen hatása, két kvantumállapotának megfelelően,
a közeli „X” mag jelét két jellé – dubletté – „hasítja fel”), s ha I
= ½ az X spinre is, akkor ugyanez történik az A jellel is. Ez az ún.
spin–spin kölcsönhatás tehát kölcsönös, s ezért a felhasadás is
azonos mértékű. Mivel a jelenség molekulán belüli természetű, a
magpolarizációt okozó B0 külső mágneses tér s ennek frekvenciája nem
befolyásolja, ezért nem függ tőle: frekvenciainvariáns.
A spin–spin kölcsönhatás tehát jelfelhasadásokhoz,
multiplicitáshoz vezet. Ha egy „A” mag több, kémiailag ekvivalens
(azonos kémiai környezetben lévő) ½ spinű „X” maggal van
kölcsönhatásban, utóbbiak kvantumállapotainak többféle variációja
lehetséges, s ezért az A jel több komponensre hasad. Ha az X magok
száma n, akkor az A jel felhasadása (n + 1)-szeres (az X jel
természetesen n-től függetlenül dublett, ha csak egy A mag vesz
részt a kölcsönhatásban). A jelintenzitások a legegyszerűbb esetben
a Pascal-háromszögből kaphatók (a binomiális együtthatóknak felelnek
meg), és a multiplettek ekvidisztáns jelekből épülnek föl. Az
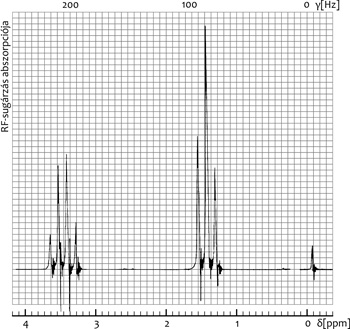
etil-klorid (CH3CH2 Cl) 1H NMR spektrumában például a metilén-jel
1:3:3:1 kvartett, a metil-jel 1:2:1 triplett (intenzitásarány 2:3),
a metil H-k négyféle és a metilén hidrogének háromféle
elrendeződésének megfelelően (2. ábra).
2. ábra
Ilyenkor a két fő paraméter könnyen megkapható a
spektrumokból: a d a szimmetrikus multiplettek középpontjaként, a J
a multiplettek bármely két szomszédos vonalának távolságaként. Az
itt leírt igen egyszerű multiplett-szerkezetek azonban csak bizonyos
feltételek fennállása esetén jelentkeznek, s gyakran ennél sokkal
bonyolultabb, egyszerűen nem áttekinthető jelrendszerek lépnek fel,
amikor a paraméterértékeket csak kvantumkémiai számításokkal
kaphatjuk meg. Igaz ugyan, hogy bizonyos korlátokkal, a bonyolult
spektrumokat a fent leírt egyszerűbb szerkezetűvé alakíthatjuk
(legegyszerűbben nagyobb B0 mágneses térerősségen működő
mérőberendezést alkalmazva).
A J-k képviselte információfajta jelentőségét az
adja, hogy a csatolási állandók nagysága a kölcsönható izotópoktól,
az azokat összekötő kémiai kötések számától és természetétől, s ami
a legfontosabb, a kölcsönható magok kölcsönös helyzetétől: a
vizsgált molekula térszerkezetétől igen érzékenyen függ. Így az NMR
a sztereokémiai problémák megoldásának rendkívül hatékony eszköze,
miközben más műszeres módszerek csak ritkán, speciális esetekben
alkalmasak térszerkezet-vizsgálatra. Kis túlzással az NMR nyitott
utat a 3D-kémia számára, kísérleti adatokat szolgáltatva a kutatók
számára a harmadik dimenzió felé. A molekulák addig síkban ábrázolt
és képzelt szerkezete kitágult térbeli, háromdimenziós alakzattá.
Önkényesen csak egy-két példát kiragadva: A 3J(H,H)
– vicinális – homonukleáris csatolási állandó lehetővé teszi a
geometriai és gyűrű- (cisz- és
|
|
|
transz-) izomerek (1,2-diszubsztituált olefinek,
diszubsztituált telített gyűrűs vegyületek, például ciklohexán-,
oxirán-származékok stb.) vagy az aromásgyűrűben különbözőképpen
(orto-, meta-, para-) helyettesített származékok, cukor-anomerek,
eltérő sztereovázas szteroidok stb. egyszerű és biztonságos
megkülönböztetését.
A csatolások kínálta elképesztő
információgazdagságot beláthatjuk a fenti – 3J(H,H) – konvencionális
jelölésből, ahol a 3 a kölcsönható magokat elválasztó/összekötő
kötések számát, a két H a kölcsönható izotópokat jelöli, ha
meggondoljuk, hogy a kötések száma 1–5, extrém eseteket is
figyelembe véve akár 1–9 is lehet, a két H bármelyike helyett pedig
bármely két azonos vagy különböző mágneses mag szerepelhet (homo-,
illetve heteronukleáris csatolások), s akkor még nem vettük
figyelembe a kötések jellegét és térbeli elrendeződését!
A spin–spin csatolások egyrészt új, gazdag
információforrást képviselnek, másrészt azonban bonyolítják, gyakran
teljesen áttekinthetetlenné teszik az NMR-spektrumokat. Ez a hátrány
azonban megszüntethető az ún. kettős rezonancia (DR – double
resonance) vizsgálatokkal. Ennek lényege, hogy a spin–spin
kölcsönhatásban lévő atommagok vagy csoportok egyikét a jelének
megfelelő RF-fel a mintát külön besugározva telítjük. Ekkor a
megfelelő magok energialeadása, emissziója felgyorsul, és ennek
következtében kvantumállapota olyan gyorsan változik, hogy azt a
vele kölcsönhatásban lévő partner már nem tudja követni. Az
eredmény, hogy az utóbbi magok, illetve csoportok jelmultiplicitása
(felhasadása) megszűnik. Tehát a DR-méréssel leegyszerűsíthetők a
spektrumok, s egyúttal újabb adatokhoz jutunk a szerkezetről. A
kettős és többszörös rezonanciának igen sok fajtája van (a fent
leírt legegyszerűbbet lecsatolásnak nevezzük), és ezek a
legváltozatosabb adatokkal gazdagítják a kémiai szerkezetről
szerezhető ismereteinket. Nemcsak a kölcsönható magpárokat,
csoportokat azonosíthatjuk, de másfajta adatok birtokába is
juthatunk: így kvantumállapotok azonosítása, más multiplettektől
fedett jelek kémiai eltolódásának pontos meghatározása stb. válik
lehetővé. Mód van egy adott magfajta összes kölcsönhatásának
megszüntetésére, a BB (broad band) lecsatolással, amikor egy adott
mag, például a hidrogén kémiai eltolódásának teljes tartományát
lefedő frekvencianyalábbal sugározzuk be a mintát, s ezzel
megszüntetjük az összes H-mag okozta felhasadást. Ezt a módszert
alkalmazzák a 13C NMR-spektrumok felvételéhez, amely így vonalas
formában regisztrálható. (A C,H-kölcsönhatások okozta felhasadások
nem jelentkeznek, a C,C-kölcsönhatások két 13C izotóp egymáshoz
közeli előfordulásának rendkívül kicsiny valószínűsége miatt szintén
nem okoznak észrevehetően felhasadt jeleket.) A vonalas spektrumban
nemcsak nagyobbak a jelintenzitások (az eredeti multiplett jeleinek
egybeesése és az alább említendő NOE miatt), de könnyebb
digitalizálhatóságuk révén egyszerűbb a számítógépes
adatfeldolgozásuk is.
Miután a csatolási állandók érzékenysége a
molekulák térszerkezetétől hármodimenziósra „tágította”
szerkezetüket, az a tapasztalat, hogy függenek a mérési
körülményektől, az addig merevnek tekintett molekulák világát
mozgásba is hozta. Azt ugyan már korábban is tudták, hogy vannak –
méghozzá igen nagy számban – nem merev, flexibilis molekulák, de a
molekuláris mozgásokat kísérletileg kimutatni, ezek változásait,
sebességét kvantitatíve is követni elsőként a dinamikus NMR – DNMR –
volt képes. A mozgékony molekulák NMR-spektrumai változnak a
hőmérséklettel: a VT- (variable temperature-) NMR, a változó
hőmérsékleteken végzett mérések betekintést engednek a különféle
atomi és molekuláris mozgások világába, ezek lejátszódására vagy
befagyására, gyorsulására vagy lassulására a csatolási állandók
térbeli változásokkal szembeni érzékenysége, illetve a folyamatok
sebességétől függő mérhetősége révén. Az atomi és gyűrűinverziók, a
különféle konformációs mozgások, a gátolt rotáció, a
vegyérték-izomerizáció és a különböző ligandcsere-folyamatok
előfordulásának igazolása, természetének tanulmányozása,
termodinamikai (aktiválási) paramétereinek kiszámítása vált lehetővé
a VT-NMR felhasználásával.
Az NMR-mérésekkel nyerhető információfajták közül
rendkívül fontos az ún. T1 (spin-rács) relaxációs idő is. A T1 a
gerjesztett magok energiaemissziójának (alapállapotba
visszatérésének) sebességét méri, és a 13C-magokra jól mérhető
nagysága az adott mag mozgási szabadságától függ: minél szabadabban,
gyorsabban mozog az adott atom, annál nagyobb T1C. A mért T1C
értékek például egy normál láncú zsíralkoholnál a
hidroxil-csoporttól távolodva folyamatosan növekszenek. Az
OH-csoportok között kialakuló hidrogénhidak ugyanis mintegy
lehorgonyozzák, rögzítik a lánc OH-t tartalmazó végét, és ettől
távolodva egyre szabadabban, gyorsabban mozognak (a szén-szén
egyszeres kötések körül forognak) a metilén-csoportok szénatomjai.
Ezt a jelenséget használják fel például az enzim-szubsztrát
kölcsönhatások rögzítési helyének meghatározására (a szubsztrát
kötőhelyének közelében lévő szénatomokra kisebb, a távolabbiaknál
fokozatosan nagyobb T1C értékek mérhetők). A patológiás sejtekben a
sejtfalhoz rendeződő szerkezetből kiszabaduló, mozgékonyabb
vízmolekulák T1H értékei megnőnek az egészséges sejtekben
mérhetőkhöz képest. A mért T1H értékek 3D-ábrázolása így a beteg
sejtek (például rosszindulatú daganat) fantomképét eredményezi. Ez
az NMR orvosdiagnosztikai alkalmazásának, az MRI (MR imaging)
módszerének alapelve.
A mag-Overhauser-effektus (NOE) (Noggle – Schirmer,
1971) az NMR-spektroszkópia azon adatforrása, amely atom–atom
távolságoknak az egykristály-diffrakciós mérésekéhez hasonló
pontosságú mérését, s ezzel óriásmolekulák, fehérjék, peptidek,
polimerek térszerkezet-meghatározását teszi lehetővé oldatban;
anélkül tehát, hogy röntgenmérésre alkalmas kristályokra lenne
szükség. A NOE lényege: ha egy magot fölös energiával gerjesztünk
(telítjük), akkor a vele valamilyen kapcsolatban lévő más mágneses
magok megoszlása a kvantumállapotok között úgy változik (nő a
betöltöttség-differencia), hogy az utóbbi mag több gerjesztő
energiát képes elnyelni az RF-sugárzásból, következésképpen nő a
jelintenzitása. (E jelenség a forrása a 13C NMR-spektrumok
jelintenzitás-növekedésének a BB-lecsatolás következtében.) Itt a
„kapcsolat” igen tágan értelmezendő: elegendő például két oldószer
keverékének egyik komponensében a H-atomokat gerjeszteni ahhoz, hogy
a másik oldószer jele a 1H NMR-spektrumban intenzívebbé váljék. Az
intenzitásnövekedés a magok távolságának hatodik (!) hatványával
fordítottan arányos, tehát a távolságmérés több nagyságrenddel
pontosabb. Ha egy „normál” és a megfelelő gerjesztett (NOE
intenzitásnövekményeket tartalmazó) spektrum különbségét képezzük (a
modern mérőberendezések számítógépének segítségével ez rendkívül
egyszerű), az így nyert DIFFNOE- vagy DNOE- (differenciál-NOE)
spektrumban csak a növekmények, az intenzívebbé vált jelek láthatók,
s ebből kiderül, hogy mely atomok vannak a gerjesztetthez térben
közel, s mekkora közöttük a távolság. Ez a modern makromolekuláris
NMR-kutatások, elsősorban a peptid- és fehérjemolekulák,
biopolimerek 3D-szerkezetmeghatározásának alapelve.
A jelintenzitások, a kémiai eltolódások és ezek
különbsége is arányos a B0 térerővel, s mivel – mint említettük –
ezek igen kicsinyek, a térerő növelése alapvető fontosságú az
NMR-mérések sikeressége és minősége szempontjából. Azt is
említettük, hogy erősebb mágneses térben egyszerűsödnek a gyengébb
tér esetén bonyolultabb spektrumok. Ezért az NMR-módszer fejlődése,
alkalmazhatósági lehetőségeinek bővülése nagymértékben az egyre
erősebb tereknek köszönhető. A kezdetben (az 1950–1960-as
évtizedekben) használatos vasmagos, permanens mágneseket 1970 táján
elektromágnesek, majd az 1980-as évektől a szupravezető mágnesek
váltották fel az NMR-spektrométerekben. Ez a H-magok
rezonanciafrekvenciájának 30–60 MHz-ről 100 MHz-re, illetve 200
MHz-ről mára már 1 GHz-re növekedését jelentette. A mérés
érzékenysége ezzel több mint egy nagyságrenddel javult, de az ún.
pulzusgerjesztés és a mérési adatoknak a Fourier-transzformáció
elvére épülő feldolgozása (PFT-NMR) további több nagyságrend
érzékenységjavulást eredményezett (Ernst et al., 1987). A kezdetben
csak a H-magok spektrumának felvételére használható műszerek így
fokozatosan alkalmassá váltak előbb csak néhány további, ½ spinű,
nagy természetes izotópgyakoriságú mag (19F, 31P), majd lassanként
egyre érzéketlenebb izotópok mérésére is. A korszerű,
számítógép-vezérelt, szupravezető mágnessel működő FT-spektrométerek
lehetővé tették a legérzéketlenebb izotópok vizsgálatát is:
beköszöntött a multinukleáris NMR-korszak.
A számítógépek teljesítőképességének növekedése és
a PFT-módszer alkalmazása egyre nagyobb adatmennyiségek egyre
gyorsabb feldolgozását tette lehetővé. Ennek köszönhetően terjedtek
el a spinrendszerek gerjesztésére alkalmazott pulzusszekvenciák,
amelyekkel ugyanazt a vizsgálati mintát besugározva más-más, igen
sokféle információ birtokába lehet jutni (Sanders – Hunter, 1987;
Derome, 1987). A spinrendszerek reagálását kezdetben egyetlen
gerjesztőfrekvencia függvényében (lineárisan) ábrázoló spektrumokat
az 1970-es évektől kiegészítették, illetve felváltották a
többgerjesztő RF-besugárzásával nyerhető 2D-, illetve sokdimenziós
regisztrátumok: beköszöntött a többdimenziós NMR-spektroszkópia
korszaka. A pulzusszekvenciákkal (több és többféle gerjesztő teret
váltakozva, kombinálva, ezeket közöttük változó tartamú szünetek
beiktatásával alkalmazva) valósággal táncra perdíthetjük a spineket,
általunk tervezett koreográfiát előírva számukra (Freeman, 1996): a
B0 tér indukálta mágneses momentumokat a legkülönbözőbb irányú
beállásokra, mozgásokra kényszerítve ugyanazon vizsgálati mintáról
szinte korlátlan számú, s a legváltozatosabb információkat
szolgáltató regisztrátumokat készíthetünk. Az alapparaméterek (d-k,
J-k, T1-ek, jelintenzitások) képviselte adatokat, s az ezekből a
vizsgált anyag szerkezetéről szerezhető információkat e mérésekkel
igen sok továbbival gazdagíthatjuk. Csak egy-kettőt említve az
ekként kapott információk közül: párosíthatjuk az egyes szénatomok
13C NMR-jelét a hozzájuk kapcsolódó hidrogének 1H jelével (s
hasonlóan bármely X atom jelét a vele kémiai kötésben lévő Y atom
jelével), megállapíthatjuk adott molekulában az atomok kapcsolódási
sorrendjét (molekulatopológia!), meghatározhatunk atom–atom
távolságokat, spin-spin kölcsönhatásban lévő csoportokat, molekulák
diffúziós sebességét és irányát, intra- és intermolekuláris
kölcsönhatásokat (például H-hidak, rétegződések kialakulását), és
igazolhatunk különféle mozgásokat (atomi és gyűrűinverziók,
ligandumcserék, vegyérték-izomerizáció, tautomer egyensúlyok és
változásaik stb.). A méréseket végző szakember fantáziája,
felkészültsége, tapasztalata függvényében az anyagokról gyűjthető
információk fajtája és száma úgyszólván határtalan, a kiegészítő
adatok gyűjtésére szolgáló méréstechnikák tárháza szinte
kimeríthetetlen!
Ma már elképzelhetetlen kémiai kutatás NMR nélkül.
De a mérések automatizálása lehetővé teszi sok minta felügyelet
nélküli folyamatos minőségellenőrzését, kémiai reakciók
lejátszódásának követését, s ekként a kutatás mellett a vegyipar
számára is nélkülözhetetlen az NMR-spektroszkópia módszer.
NMR-spektrométerek tucatjai működnek nemcsak a nagy
kutatócentrumokban, de az ipartelepeken is. Valóságos
NMR-városrészek létesültek, például Japánban több száz műszerrel és
azokat kiszolgáló több ezres mérőszemélyzettel. A fejlődés ma sem
állt meg, egyedül a mérések anyagigénye terén is lenyűgöző az
előrelépés. Túlzás nélkül állítható: a kémiai tudomány történetének
NMR-korszakát éljük!
Kulcsszavak: mágneses magmomentum, rezonanciaabszorpció, kémiai
eltolódás, spin-spin csatolás, kettős rezonancia, mérés változó
hőmérsékleten, spin-rács relaxáció, mag-Overhauser-effektus,
Fourier-transzformációs NMR, pulzusgerjesztés, szupravezető
mágnesek, két- és többdimenziós NMR, ipari alkalmazás
IRODALOM
Breitmaier, Eberhard – Voelter, Wolfgang
(1974): 13C NMR Spectroscopy. Verlag Chemie, Weinheim
Derome, Andrew E. (1987): Modern NMR
Techniques for Chemistry Research. Pergamon Press, Oxford
Ernst, Richard R. – Bodenhausen, G. –
Wokaun, A. (1987): Principles of Nuclear Magnetic Resonance in One
and Two Dimensions. Clarendon Press, Oxford
Freeman, Ray (1996): Spin Choreography.
Spectrum Academic Publishers, Oxford
Hore, Peter J. (1995): Mágneses
magrezonancia. Nemzeti Tankönyvkiadó, Budapest
Noggle, Joseph H. – Schirmer, Roger E.
(1971): The Nuclear Overhauser Effect. Academic Press, New York
Sanders, Jeremy Keith Morris – Hunter,
Brian Keith (1987): Modern NMR Spectroscopy. A Guide for Chemists.
University Press, Oxford
Sohár Pál (1976): Mágneses magrezonancia
spektroszkópia I–II. Akadémiai, Budapest
Sohár Pál (1983a): Nuclear Magnetic
Resonance Spectroscopy. I–III. CRC Press, Boca Raton, Florida
Sohár Pál (1983b):
Szénrezonancia-spektroszkópia. (A kémia új eredményei 59) (szerk.
Csákvári Béla), Akadémiai, Budapest
Wehrli, Felix W. - Wirthlin, Toni (1976):
Interpretation of Carbon-13 NMR Spectra. Heyden, London
|
|
